Tarantula toxin attacks with molecular stinger
Venom immobilizes prey by interfering with sodium channels that generate electrical signals in the animal’s nerve cells.Media Contact: Leila Gray - 206.475.9809, leilag@uw.edu

Oversized, hairy tarantulas may be unsightly and venomous, but surprisingly their hunter toxin may hold answers to better control of chronic pain.
A bird-catching Chinese tarantula bite contains a stinger-like poison that plunges into a molecular target in the electrical signaling system of their prey’s nerve cells.
A new high-resolution cryo-electron microscopy study shows how the stinger quickly locks the voltage sensors on sodium channels, the tiny pores on cell membranes that create electrical currents and generate signals to operate nerves and muscles. Trapped in their resting position, the voltage sensors are unable to activate.
The findings are published Nov. 23 in Molecular Cell, a journal of Cell Press.

“The action of the toxin has to be immediate because the tarantula has to immobilize its prey before it takes off,” said William Catterall, professor of pharmacology at the University of Washington School of Medicine. He was the senior researcher, along with pharmacology professor and Howard Hughes Medical Institute investigator, Ning Zheng, on the study of the molecular damage inflicted by tarantula venom.
While some might dismiss those tarantulas as ugly, tough and mean, medical scientists are actually interested in their venom’s ability to trap the resting state of the voltage sensor on voltage-gated sodium channels and shut them down. Such studies of toxins from these “big, nasty dudes,” as Catterall describes them, could point to new approaches to structurally designing drugs that might treat chronic pain by blocking sensory nerve signals.
Catterall explained that chronic pain is a difficult-to-treat disorder. Efforts to seek relief can sometimes be a gateway to opiate overdose, addiction, prolonged withdrawal, and even death. The development of safer, more effective, non-addictive drugs for pain management is a vital need.
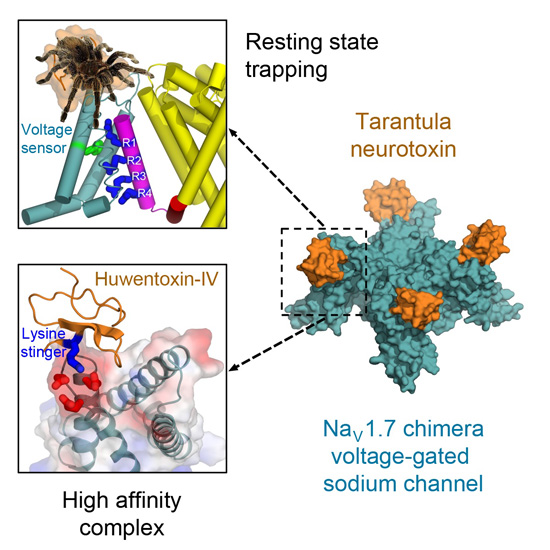
However, because it has been hard to capture the functional form of the tarantula toxin-ion channel chemical complex, reconstructing the toxin’s blocking method in a small molecule has so far eluded molecular biologists and pharmacologists seeking new ideas for better pain drug designs.
Researchers overcame this obstacle by engineering a chimeric model sodium channel. Like mythical centaurs, chimeras are composed of parts of two or more species. The researchers took the toxin-binding region from a specific type of human sodium channel that is crucial for pain transmission and imported it into their model ancestral sodium channel from a bacterium. They were then able to obtain a clear molecular view of the configuration of the potent toxin from tarantula venom as it binds tightly to its receptor site on the sodium channel.
This achievement revealed the structural basis for voltage sensor trapping of the resting state of the sodium channel by this toxin.
“Remarkably, the toxin plunges a ‘stinger’ lysine residue into a cluster of negative charges in the voltage sensor to lock it in place and prevent its function,” Catterall said. “Related toxins from a wide range of spiders and other arthropod species use this molecular mechanism to immobilize and kill their prey.”
Catterall explained the medical research importance of this discovery. The human sodium channel placed into the chimeric model is called the Nav1.7 channel. It plays an essential role, he noted, in transmission of pain information from the peripheral nervous system to the spinal cord and brain and is therefore a prime target for pain therapeutics.
“Our structure of this potent tarantula toxin trapping the voltage sensor of Nav1.7 in the resting state,” Catterall noted, “provides a molecular template for future structure-based drug design of next-generation pain therapeutics that would block function of Nav1.7 sodium channels.”
The lead authors on the study were Goragot “George” Wisedchaisri and Lige Tonggu, both of the UW School of Medicine Department of Pharmacology. Tamer M. Gamal El-Din, also of Pharmacology, and Eedann McCord, now with the Department of Physiology and Biophysics at the UW medical school, contributed to the research.
The research was supported by National Institutes of Health research grants R01 NS015751, R35 NS111573 and R01 HL112808, NIH training grant T32 GM007750 and by the Howard Hughes Medical Research Institute.
The researchers declared no competing interests
For details about UW Medicine, please see our About page.