Patterns in DNA reveal hundreds of unknown protein pairings
Method will be applied to human genome to learn more about how proteins interact inside our cells to carry out biological functionsMedia Contact: Leila Gray, 206.685.0381, leilag@uw.edu
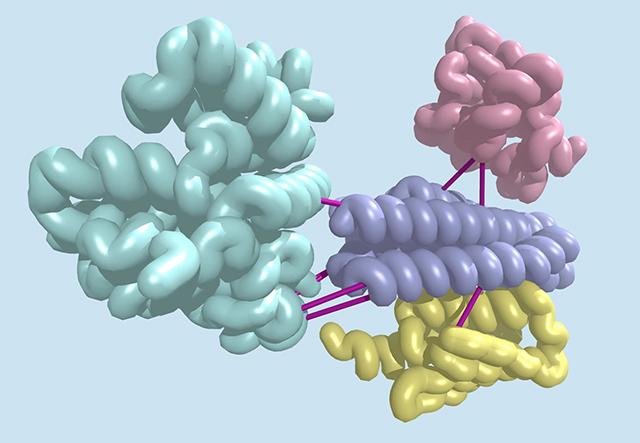
Sequencing a genome is getting cheaper, but making sense of the resulting data remains hard. Researchers have now found a new way to extract useful information out of sequenced DNA.
By cataloging subtle evolutionary signatures shared between pairs of genes in bacteria, the team was able to discover hundreds of previously unknown protein interactions. This method is now being applied to the human genome, and could produce new insights into how human proteins interact.
The project is a collaboration between scientists at the University of Washington School of Medicine and Harvard University. Their report appears in the July 11 issue of Science. A computational biology perspective on the work also appears in Science.
“Protein–protein interactions are fundamental to biological function. It’s remarkable that they can now be predicted en masse using the large amounts of genomic sequence data that have been generated in recent years,” said senior author David Baker, professor of biochemistry at the University of Washington School of Medicine.
Cells are packed with proteins, many of which must physically interact in order to function. This can mean coming together to copy DNA or to form long fibers like those found in muscle. In many cases, however, scientists still do not know which proteins interact. Discovering new pairings can be slow, laborious, and costly.
Looking for a better way, a team of four computational biologists studied a phenomenon called co-evolution, wherein changes in one gene are associated with changes in another. This can indicate that two genes are linked in some important way.
For example, if one gene mutates to produce a protein with an altered shape, a second may evolve to produce a protein with a shape complementary to the first, thereby preserving the ability of the two proteins to interact.
In recent years, researchers have found evidence for some of these subtle molecular interactions in an organism's DNA.
“Co-evolution has been useful for understanding how specific proteins interact, but we can

now use it as a tool for discovery,” said lead author Qian Cong, a postdoctoral fellow at the UW School of Medicine.
The research team compared more than 4,000 genes from E. coli to DNA sequences from more than 40,000 other bacterial genomes. This large stockpile of genetic information allowed the researchers to use a bespoke statistical model to assess co-evolution between each E. coli gene.
After several rounds of analysis, 1,618 pairs were found to have the strongest evidence of co-evolution. By comparing their results to a small set of already characterized protein–protein interactions, the researchers achieved considerably higher accuracy than previous experimental screening methods.
Among the newly discovered interactions were a few that hint at new biological insights. One of these, an interaction between a protein toxin and its antitoxin, may help explain, the researchers speculate, why some E. coli dominate their microbial niche. Another newfound pairing suggests that a protein called PstB, which was known to play a role in metabolism, may also help coordinate protein synthesis and mineral transport.
“It is rare in biology for a software tool to make predictions that are promising enough to test, but that is exactly what’s happening here,” said Cong. There are literally hundreds of follow-up experiments that could be performed in labs around the world.”
The team also scoured the genome of Mycobacterium tuberculosis, a disease bacterium distantly related to E. coli. They identified 911 protein–protein interactions with high confidence. 95 percent of these had never been previously described. Seventy involve proteins that may contribute to the virulence of M. tuberculosis, the researchers report. These findings may open new routes to develop drugs against the deadly pathogen.
“We are going to apply this tool to more pathogens, and the human genome,” says Cong. “Our success will depend on how much work other scientists put into annotating which parts of the genome are genes and which parts are something else.”
This project was funded by the Washington Research Foundation, National Institute of General Medical Sciences (R01-GM092802-07), National Institute of Allergy and Infectious Diseases (HHSN272201700059C), Office of the Director of the National Institutes of Health (DP5OD026389), Spark Therapeutics, and IPD Director’s Fund from Bruce and Jeannie Nordstrom. Cong is a Washington Research Foundation Innovation Fellow.
This research used resources of the National Energy Research Scientific Computing Center.
The authors declare no competing interests.
Written by Ian Haydon of the Institute for Protein Design.
For details about UW Medicine, please see our About page.