New human reference genomes reveal greater diversity
Data includes individuals from around the world to better capture complexities of human genetic variation.Media Contact:
- UW Medicine: Leila Gray, 206.475.9809, leilag@uw.edu
- Jackson Laboratory: Sarah Laskowski, 860.837.2120, sarah.laskowski@jax.org
- Heinrich Heine University Düsseldorf: Arne Claussen, Arne.Claussen@hhu.edu
- European Molecular Biology Laboratory Heidelberg: Mathias Jäger, mathias.jaeger@embl.org

Exactly 20 years after the successful completion of the Human Genome Project, an international group of researchers, the Human Genome Structural Variation Consortium, has now sequenced 64 human genomes at high resolution. This reference data includes individuals from all over the world to better capture the genetic diversity of the human species. Among other applications, the work enables population-specific studies on genetic predispositions to human diseases as well as the discovery of more complex forms of genetic variation, as the 65 authors report in the Feb. 26 issue of the scientific journal Science.
Please see EurekAlert news release on this paper.
In 2001, the International Human Genome Sequencing Consortium announced the first draft of the human genome reference sequence. The Human Genome Project, as it was called, had taken more than eleven years of work and involved more than 1,000 scientists from 40 countries. This reference, however, did not represent a single individual but instead was a composite of humans, and that approach could not accurately capture the complexity of human genetic variation.
Building on this draft, scientists have carried out many sequencing projects over the past 20 years to identify and catalog genetic differences between an individual and the reference genome. Those differences usually focused on small single base changes and missed larger genetic alterations. For the first time, current technologies are beginning to detect and characterize larger differences – called structural variants – such as insertions of several hundred letters. Structural variants are more likely than smaller genetic differences to interfere with gene function.
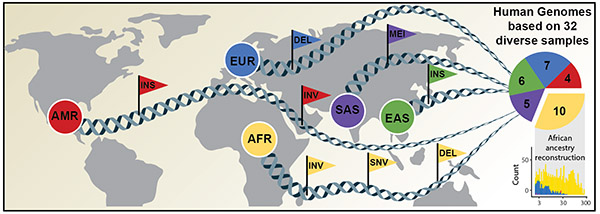
The international research team’s article in Science announces a new, considerably more comprehensive reference dataset obtained using a combination of advanced sequencing and mapping technologies. The new reference dataset includes 64 assembled genomes representing 25 different human populations from across the globe. Importantly, each of the genomes was generated without guidance from the first human genome and as a result better captures genetic differences from these diverse populations.
The study was led by scientists from the Department of Genome Sciences, University of Washington School of Medicine in Seattle, The Jackson Laboratory for Genomic Medicine in Farmington, Conn., European Molecular Biology Laboratory Heidelberg and the Heinrich Heine University Düsseldorf.
“With these reference data, individual differences in terms of various types of genetic variants can now be studied with unprecedented accuracy," emphasized the first author of the study, Peter Ebert from the Institute of Medical Biometry and Bioinformatics at Heinrich Heine University. This is because the distribution of genetic variants can differ significantly between populations. This is due to the fact that spontaneous changes in the genetic material occur continuously. If these mutations are not a hindrance to survival, they can be passed on and consolidated in a population group.
“Each of these individual genomes is being resolved more completely for a fraction of the price of the first human genome,” commented senior author, Evan Eichler, professor of genome sciences, University of Washington School Medicine, who was also a member of the original Human Genome Project. “We are discovering remarkable differences in genomic organization which have been missed until now. Understanding these differences improves our ability to make genetic discoveries related to health and disease especially in groups that have been traditionally underserved by genomics research."
Peter Audano, a bioinformatics specialist in the Eichler lab and a lead author of the paper, added, “The technology we have today can see into blind spots that have hidden information about diseases and our history.” The effort resolved more than 100,000 structural variants most of which were previously unknown.
The new reference data begin to resolve the full spectrum of genetic variants needed to improve association with human genetic diseases. The aim is to estimate the individual risk of developing certain genetic diseases and to understand the underlying molecular mechanisms. This information, in turn, can be used as a basis for more targeted therapies and preventative medicine.
Eichler noted, “The approach is fundamentally changing the way we discover genetic variation. We fully resolve the paternal and maternal genomes of each individual first and then discover variants by comparing the assembled genomes to other references instead of aligning sequence reads to a composite reference.” He believes that this “is a game changer and will lead to many more genetic discoveries in the future.”
“It won’t happen tomorrow,” he added, “but this is the way all human genomes will be sequenced clinically in the future. Someday each person will have their individual human genome project to call their own and having that information will improve their health."
This study builds on a new method published by these researchers last year in Nature Biotechnology to accurately reconstruct the two components of a person's genome – one inherited from a person’s father, one from the person’s mother. When assembling a person’s genome, this method eliminates the potential biases that could result from comparisons with an imperfect reference genome.
The Feb. 26 Science paper is titled Haplotype-resolved diverse human genomes and integrated analysis of structural variation.
The other lead authors on the paper are Qihui Zhu of the Jackson Laboratory and Bernando-Rodriguez Martin of EMBL. The senior and corresponding authors also include Tobias Marshal of Heinrich Heine University, Jan Korbel of EMBL, and Charles Lee of the Jackson Laboratory.
A full list of the 65 authors and their institutions is available in the paper.
Please also see the institutions' joint release on EurekaAlert.
###
Funding for this research came from: National Institutes of Health (U24HG007497, R01HG002898, R01HD0812561, R01HG007068-01A1, R01HG002385, R01MH115957, R15HG009565, 1U01HG010973, NIH 1R35GM138212, 1OT3HL147154), NIH/National Human Genome Research Institute Pathway to Independence Award (K99HG011041), German Research Foundation (391137747 and 395192176), European Research Council (773026. 716290), German Federal Ministry for Research and Education (BMBF 031L0184 and BMBF 031L0181A), Spanish Ministry of Economy, Industry and Competitiveness (SAF2015-66368-P), Wellcome Trust (WT085532 and WT104947/Z/14/Z), European Molecular Biology Laboratory, National Science Foundation of China (32070663, 61702406), National Key R&D Program of China (2017YFC0907500, 2018YFC091040, 2018ZX10302205 ), BMBF-funded de.NBI Cloud within the German Network for Bioinformatics Infrastructure 39 (031A537B, 031A533A, 031A538A, 031A533B, 031A535A, 031A537C, 031A534A, 031A532B), Howard Hughes Medical Institute, European Research Council, Ewha Womans University, The First Affiliated Hospital of Xi’an Jiaotong University, Centers for Common Disease Genomics, and National Human Genome Research Institute (UM1HG008901).
For details about UW Medicine, please see our About page.