Molecular atlases reveal how human cells develop and grow
The two atlases map gene expression and availability as cells differentiate into different cell types and tissues.Media Contact: Leila Gray - 206.475.9809, leilag@uw.edu
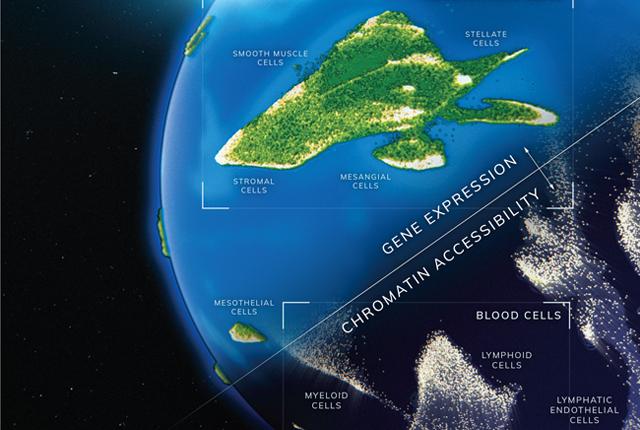
UW Medicine researchers at the Brotman Baty Institute in Seattle have created two cell atlases that track gene expression and chromatin accessibility during the development of human cell types and tissues.
One atlas maps gene expression within individual cells across 15 fetal tissues. The second atlas maps the chromatin accessibility of individual genes within the cells.
Together, the atlases provide a fundamental resource for understanding gene expression and chromatin accessibility in human development and are unprecedented in scale. The techniques reported in the two papers make it possible to generate data on gene expression and chromatin accessibility for millions of cells.
The atlases are described in back-to-back publications in the November 13 issue of the journal Science. In addition to the BBI and UW Medicine, other collaborators from Illumina, Inc., University of Arizona, the Fred Hutchinson Cancer Research Center, the Max Planck Institute for Molecular Genetics and the University of Rochester Medical Center contributed to these studies.
Gene Expression Atlas
Gene expression is the process in which a cell uses the instructions stored in its DNA to direct protein synthesis. These proteins, in turn, determine a cell’s structure and function. The gene expression atlas maps where and when gene expression occurs in different cell types as they grow and develop.
“From these data, we can directly generate a catalog of all major cell types across human tissues, including how those cell types might vary in their gene expression across tissues,” said lead author, Junyue Cao, PhD. Cao completed this work as a postdoctoral fellow in the laboratory of Jay Shendure,professor of genome sciences at the University of Washington School of Medicine, a Howard Hughes Medical Institute investigator, and scientific director of the Brotman Baty Institute. Cao is now an assistant professor at Rockefeller University.
“There is an overarching goal of the field to profile the genetic programs that are present in a human at as broad of a scale, and with as much resolution, as possible,” said Shendure.
To create the atlas, the researchers profiled gene expression across 15 types of fetal tissue by using a technique called sci-RNA-seq3. This technique labels each cell with a unique combination of three DNA "barcodes," thereby allowing the researchers to keep track of cells without physically separating them.
Once they obtained the sequences. the scientists used computer algorithms to recover the single cell information, cluster cells by type and subtype, and identify their developmental trajectories. The scientists profiled over 4 million single cells and identified 77 main cell types and approximately 650 cell subtypes.
They also compared the atlas to an existing atlas of mouse embryonic development. Co-senior author Cole Trapnell, associate professor of genome sciences at the UW School of Medicine and a Brotman Baty Institute scientist, explained, “When we combine this data with previously published data, we can directly delineate the cell’s developmental path for all major cell types.”
DNA Accessibility Atlas
The second atlas, of DNA accessibility, maps material called chromatin that allows DNA to be tightly packed into the cell nucleus. Chromatin can be open and accessible”to the molecular machinery that reads the genetic instructions encoded in the DNA or closed and “naccessible. Knowing regions of DNA that are open and closed can tell how a cell chooses to turn genes on and off.
"Studying chromatin gives you a sense of the regulatory 'grammar' of the cell,” said co-senior author Darren Cusanovich, previously a postdoctoral fellow in the Shendure lab and now an assistant professor at the University of Arizona. “The short stretches of DNA that are open, or accessible, are enriched for certain ‘words,’ which are, in turn, the basis for the cell to specify that it wants certain genes on.”
To profile DNA accessibility in individual cells, the scientists developed a new method, called sci-ATAC-seq3. Like sci-RNA-seq3, this technique also uses three different DNA “barcodes” in each cell to tag and track individual cells. However, rather than identifying all the currently expressed sequences, sci-ATAC-seq3 captures and sequences open chromatin sites.
The scientists generated nearly 800,000 single-cell chromatin accessibility profiles at ~1 million sites across 15 fetal tissues in this study. They asked which proteins were likely to interact with accessible DNA sites in each cell and how those interactions can explain the cell type. This analysis defines the control switches for development within the genome. They also identified sites of chromatin accessibility that might be associated with diseases.
“This tells us what part of the genome might be functional. We still do not know what percentage of the genome that doesn’t encode for genes can be involved in gene regulation. Our atlas now provides that information for many cell types,” said Silvia Domcke, a co-lead author of the accessibility atlas paper and a postdoctoral fellow in the Shendure lab. The other lead authors of the DNA accessibility study are Andrew Hill, previously a computational biologist in the Shendure lab and now a scientist at 10x Genomics, and Riza Daza, a research scientist in the Shendure lab.
Learn more in the Science publications, “A Human Cell Atlas of Fetal Gene Expression,” and “A Human Cell Atlas of Chromatin Accessibility.” Explore the data at https://descartes.brotmanbaty.org/
Components of these studies were funded by the Brotman Baty Insitute, Paul G. Allen Frontiers Foundation, Chan Zuckerberg Initiative, Howard Hughes Medical Institute, and National Institutes of Health.
News release written by Cognition Studio, Inc., and Michael McCarthy.
Animation created by Dani Bergey and Inessa Stanishevskaya of Cognition Studios, Inc.
NEWS MEDIA RESOURCE: Download the animation
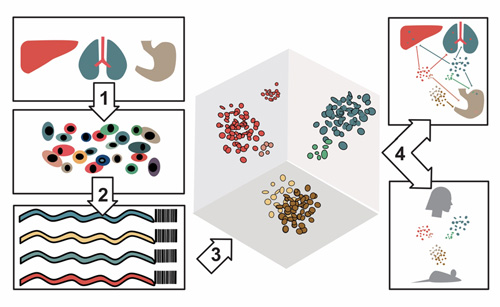
Legend for the schematic
- Cells were obtained 15 different human organs.
- In the gene expression study, mRNA transcripts were sequenced to identify which genes were being expressed. In the chromatin accessibility study, open regions of the genome were sequenced to identify which regulatory regions were active. In both cases, the RNA or the DNA were “barcoded” so their cell of origin could be tracked.
- Computer algorithms helped cluster the cells by their gene-expression or their chromatin accessibility profiles.
- The profiles of cell types were compared across their organs of origin as well as with existing databases of gene expression and chromatin accessibility across different developmental stages of mice and humans. This comparison facilitates the use of these atlases in the study of normal and abnormal development and disease.
For details about UW Medicine, please see our About page.