An atomic view of the trigger for the heartbeat
Media Contact: Leila Gray, 206.685.0381, leilag@uw.edu
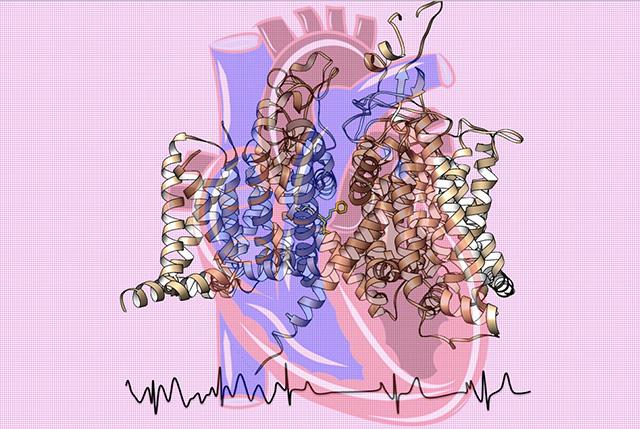
Atomic-level studies of the architecture of tiny sodium channel proteins, critical to generating electrical signals that start off each beat of the heart, are imparting striking details about their function, malfunctions, disruption by many disease mutations, and response to medication.
This structural information could become the basis for developing better diagnostics and drugs for life-threatening heart rhythm problems, according to the researchers from the University of Washington School of Medicine working in this area.
Their latest findings appear Dec. 19 in Cell in the paper, “Structure of the Cardiac Sodium Channel.” The senior authors are William Catterall and Ning Zheng, both UW School of Medicine professors of pharmacology. The first authors are Daohau Jiang and Hui Shi, UW postdoctoral fellows in pharmacology.
“The cardiac sodium channel not only initiates the heartbeat, mutations in it also cause deadly arrhythmias, and antiarrhythmic drugs act directly on it to control cardiac rhythms,” explained Catterall.
The heart is both a plumbing and electrical marvel. For each heartbeat, electrical waves travel across a healthy heart in a pattern that controls its filling and pumping in a tightly coordinated manner. The rate at which the impulse is propagated through the heart tissue relies on actions taking place at the molecular level in tiny protein pores present in cardiac cell membranes.
Sodium ions – a type of charged particles -- pass through these protein passageways in the membrane boundary between the outside and inside of the cell.

The activation and quick inactivation of these voltage-gated sodium channels are part of a series of electrical and physiological events that maintain a steady heartbeat.
“Sodium channels operate in concert with calcium channels and potassium channels to drive the heartbeat at a consistent frequency for our entire lives,” Zheng noted.
When sodium channels don’t work properly, the heart can be in trouble, even to the point of having dangerously fast and uncoordinated contractions that are life-threatening, the researchers explained. Specifically, the NaV (Latin abbreviation for sodium, V for voltage) 1.5 channel has such an indispensable role that certain mutations in those channels can be fatal, because other sodium channels in the heart cannot compensate for their loss. These mutations can cause dangerous arrhythmias in adults and even sudden death in children and young athletes.
Fortunately, many heart rhythm disturbances can be treated with drugs that block cardiac sodium channels. For example, as UW Medicine physician Michael Lenaeus, a UW assistant professor of medicine, Division of General Internal Medicine, and a co-author of the study, noted, atrial fibrillation, or “A-fib,” is increasingly prevalent among older Americans. This condition can often be treated effectively with the drug flecainide. In their recent study, the researchers sought to learn, among other things, how drugs like flecainide act within the predominant form of sodium channels found in cardiac cells.
To obtain a high-resolution, 3-D map of the channels, the scientists used advanced cryo-electron microscopy at the new Beckman Center for Cryo-EM at the UW. They wanted to explore important structural features of these sodium channels and relate their configuration to their actions in normal physiological function, dysfunction, disease mutations, toxin sensitivity and the pharmacology of antiarrhythmic drugs.
According to the scientists, their experiments provide a blueprint for understanding many various aspects of cardiac sodium channels.
Among the key findings from this work were:
- A description of some of the characteristics of the NaV1.5 channel that distinguish it from other sodium ion channels found in heart cells, as well as in nerve and muscle cells.
- Structural insights into the mode of action of various arrhythmia mutations. There are many parts of the sodium channel protein that can be disrupted by mutations. Some mutations interfere with the activation or inactivation of the sodium channel. Others cause altered function by creating a channel protein hole that leaks sodium continuously. Still other mutations, found in Brugada syndrome, can impair or block the conductance of sodium ions.
- Chemical insights into how the heart rhythm drug flecainide targets the inner pocket of the ion channel and plugs the central pore that conducts sodium ions.
- The structural determinants that distinguish tetrodotoxin sensitive and insensitive sodium channels. Tetrodotoxin, the infamous, deadly poison in pufferfish, is a sodium channel blocker that has a high affinity for sodium channels in nerves and muscles, but not for those in heart cells.
- A comparison of the conformation of an important component of the sodium ion channels that “senses” the voltage across the cell membrane and drives the channels from resting to activated states. After their rapid activation, cardiac sodium channels inactivate within 1 to 2 milliseconds, a timing that is essential to regular heart rhythm. The researchers were able to determine how mutations can upset activation of the channel or even abolish its required rapid inactivation in a variety of ways.
In their conclusion, the researchers noted that, overall, the detailed, high-resolution structures they were able to obtain for the cardiac NaV1.5 channel allowed them to elucidate the molecular basis for several
of its specific, distinctive, vital functions and to provide key chemical information for design and development of safer and more effective antiarrhythmic medications.
“Our high-resolution cryoEM structure of this iconic sodium channel gives new structure/function insights, reveals the molecular mechanisms of many inherited arrhythmias, and elucidates the exact binding site and blocking mode of the antiarrhythmic drug flecainide, which is used to treat atrial fibrillation, an increasingly prevalent problem in our aging population,” Catterall said in summarizing the results.
The research was funded by the National Institutes of Health (R01HL112808) and the Howard Hughes Medical Institute.
For details about UW Medicine, please see our About page.